Significant contributions in this field, especially for the treatment of stroke, have been made by Josef Šivic's team from the CTU CIIRC, especially by Anton and Roman Bushuiev, in collaboration with Stanislav Mazurenko's team from the Loschmidt Laboratories of Masaryk University (MU) and the International Clinical Research Centre (ICRC) and Tomáš Pluskal's team from the Institute of Organic Chemistry and Biochemistry (IOCB) of the CAS. Their new method, called PPIformer, uses machine learning to more efficiently design proteins with improved interaction properties, paving the way for significantly accelerating the development of new drugs for stroke and other diseases. The researchers presented their research results at one of the world's most important machine learning conferences, ICLR 2024, in Vienna in early May.
Unlike traditional drugs, biotherapeutics can work with the immune system or other biological processes to recognise and treat disease. These are often enzymes, antibodies or even cells, but require modification to improve biological efficacy and stability.
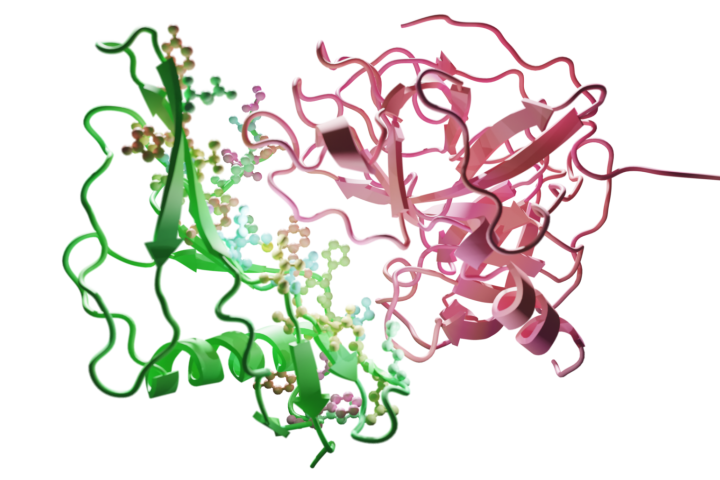
Scientists have focused on developing an improved version of the staphylokinase protein, which has already proved useful in dissolving blood clots in thrombolytic drugs given for stroke. Strokes can have serious and long-term consequences, including paralysis, speech and language problems, memory loss and emotional difficulties. Its effects can vary greatly depending on the severity of the stroke and the speed of treatment given. However, the widespread clinical use of staphylokinase is currently limited by its weak interaction with plasmin, a protein present in human blood.
"We focused on improving staphylokinase by exchanging its specific building blocks - amino acids that are responsible for these interactions," explains Josef Šivic from CIIRC CTU and adds: "In order to identify the amino acids best suited for such replacements, we trained a computational neural model that is able to learn from a large number of examples, similar to what is already common in other fields, for example in natural language processing systems such as ChatGPT. The system can predict the effect of such a change, i.e. how changes in amino acids will subsequently affect interactions between proteins."
There are millions of possible ways to modify the amino acids of proteins. "The traditional way of finding the most suitable modifications to improve drug interactions would require extensive and time-consuming experiments," points out Tomáš Pluskal from the Institute of Organic Chemistry
and Biochemistry of the CAS. Identifying these amino acid substitutions is of great practical importance for other tasks related to protein interactions, such as the design of new vaccines and biosensors.
The proposed method, called PPIformer, allows to predict the effects of amino acid substitutions on protein-protein interactions (PPIs) in a fraction of a second. The PPIformer method is based on machine learning that implements so-called "self-supervised" learning, where the model is able to learn from the data itself without the need for additional annotations.
"This means that PPIformer does not require expensive and time-consuming laboratory experiments. Instead, it relies on our newly collected and currently the largest protein-protein interaction data set obtained from publicly available protein structures," explains Stanislav Mazurenko of the Loschmidt Laboratories at MU.
PPIformer was first trained to predict amino acids masked in known protein-protein interaction structures. The principle is similar to large-scale language models, such as ChatGPT, which are trained on predicting words in sentences.
"After learning from millions of automatically generated training examples of masked amino acids, the model was retrained on a small dataset containing amino acid substitutions annotated from laboratory-measured data," says PhD student and lead author of the method Anton Bushuiev from CTU CIIRC.
The developed method, described in the paper „Learning to design protein-protein interactions with enhanced generalization”, has shown high potential in identifying beneficial mutations in staphylokinase and also in the human SARS-CoV-2 coronavirus antibody.
Designs of improved staphylokinase are currently being experimentally verified in the Loschmidt Laboratories of MU and in the International Clinical Research Centre of St. Anne's University Hospital in Brno and the Faculty of Medicine of MU. In addition, the team, in collaboration with the International Neurodegenerative Disorders Research Centre (INDRC), will extend this approach to biomolecules involved in neurodegenerative diseases.
The newly developed machine learning method was accepted at ICLR 2024 (The Twelfth International Conference on Learning Representations). ICLR is one of the top three conferences in machine learning (along with NeurIPS and ICML) and is one of the top ten most influential journals and conferences in all areas of science according to Google Scholar.
Expert Paper: A. Bushuiev, R. Bushuiev, P. Kouba, A. Filkin, M. Gabrielova, M. Gabriel, J. Sedlar, T. Pluskal, J. Damborsky, S. Mazurenko, J. Sivic, Learning to design protein-protein interactions with enhanced generalization, International Conference on Learning Representations (ICLR), 2024. https://doi.org/10.48550/arXiv.2310.18515